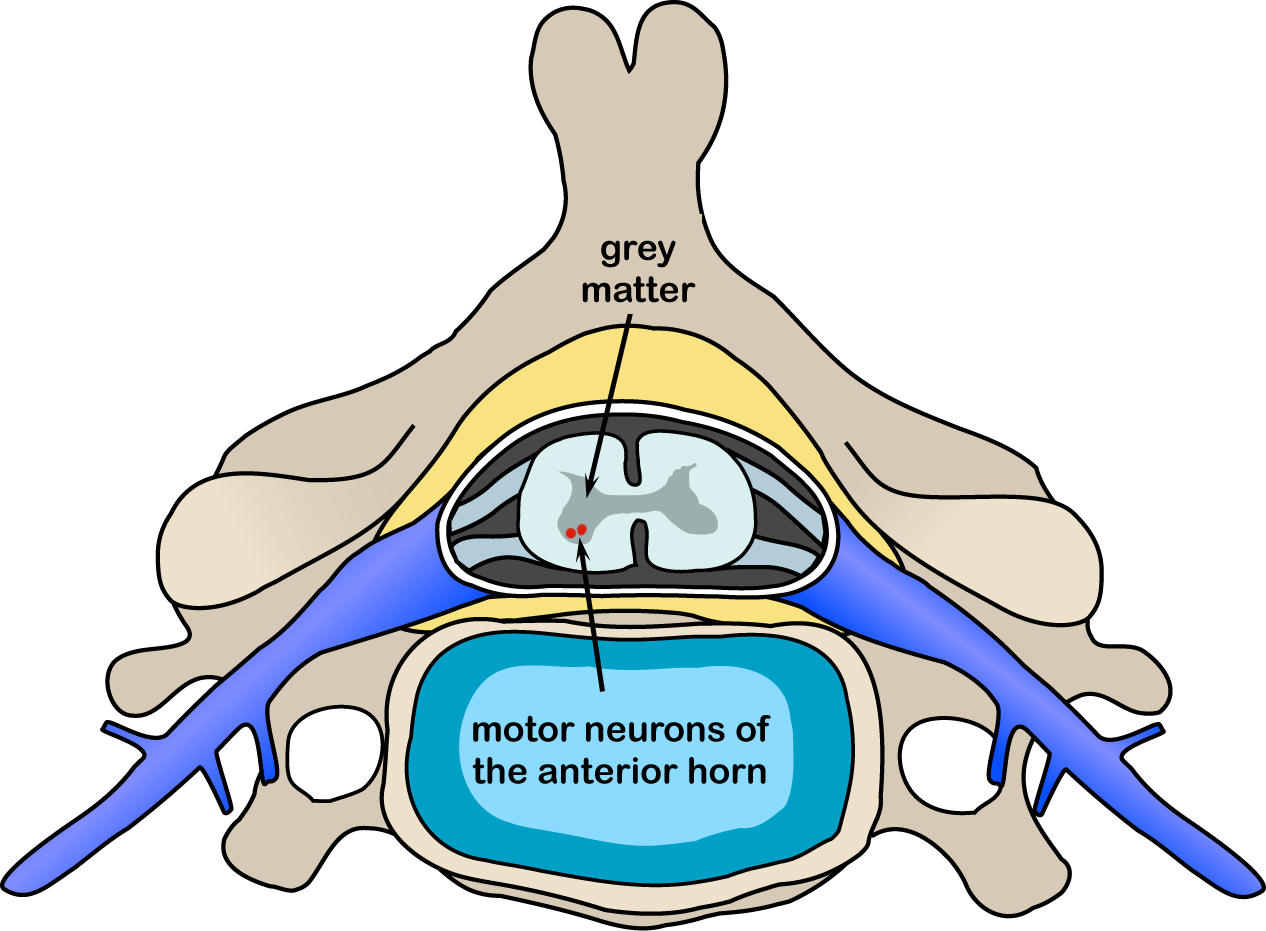
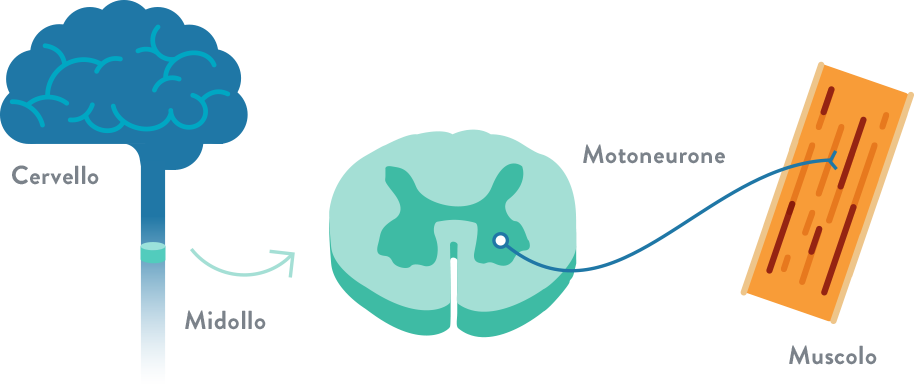
Atrofia Muscolare Spinale (SMA). Le atrofie muscolari spinali (SMA) sono un gruppo di malattie ereditarie in cui vengono colpite le cellule nervose delle corna anteriori del midollo spinale, da cui partono i nervi diretti ai muscoli. I sintomi delle SMA si manifestano pertanto a livello dei muscoli volontari. I primi sintomi comprendono debolezza muscolare nei muscoli più vicini al tronco, per poi progredire più lontano, rendendo difficile compiere attività quali andare a carponi ("gattonare"), camminare, controllare il collo e la testa e deglutire. Nei casi più gravi rende difficile la respirazione. I sensi e le percezioni sono normali, così come lo è l'attività intellettiva. La malattia ha un incidenza di 1 su 10.000 nati vivi e la frequenza dei portatori è di circa 1 su 50. In base all'età d'esordio e alla gravità della malattia, sono stati definiti quattro sottotipi: SMA di tipo 1 (malattia di Werdnig-Hoffmann), la forma più grave, con esordio prima dei 6 mesi di vita. In genere la malattia progredisce in modo piuttosto rapido e purtroppo in molti casi può portare a morte per insufficienza respiratoria o infezioni broncopolmonari. SMA di tipo 2 (forma cronica infantile), con esordio tra i 6 e i 18 mesi di vita e a progressione più lenta. La gravità e la sopravvivenza dei malati sono variabili e difficilmente prevedibili. SMA di tipo 3 (malattia di Kugelberg-Welander), con esordio tra l'infanzia e l'adolescenza. Viene denominata anche atrofia muscolare spinale benigna perché l'insorgenza è tardiva e la malattia progredisce in modo lento; rispetto alle altre forme la prognosi è quindi meno grave. Nella sua forma di gran lunga prevalente, l'Atrofia Muscolare Spinale è una malattia autosomica recessiva, perciò gli individui malati possono nascere solo se entrambi i genitori sono portatori (spesso asintomatici) dell'anomalia genetica. Per una coppia di genitori portatori sani, vi è un rischio del 25% di generare un bambino malato, indipendentemente dal sesso; il 50% di possibilità di avere un bambino o una bambina sani ma portatori; il 25% di possibilità di avere un figlio o una figlia sani e non portatori. Il 97% circa delle SMA è causato da delezioni in omozigosi dell’esone 7 del gene SMN1 (5q12.2-q13.3), che codifica per la proteina SMN (survival motor neuron), che sembra avere un ruolo nelle funzioni del nucleo cellulare, specialmente nelle cellule nervose. La diagnosi si basa sulla obiettività clinica e può essere confermata dall'analisi genetica. Possono essere utili anche l'elettromiografia e la biopsia muscolare.
Il test genetico viene effettuato mediante l’analisi MLPA (Multiple Ligation-dipendent Probe Amplification). L’MLPA è un metodo di PCR multipla che permette di rilevare il numero di copie di più frammenti (fino a 50) di DNA genomico. Il test viene impiegato per valutare lo stato di portatore di SMA a partire da piccole quantità di DNA estratto da campioni di sangue venoso periferico o, dove richiesto, da materiale fetale. In seguito all’analisi è possibile rilevare il numero di copie degli esoni 7 e 8 dei geni SMN1 e, nel caso di delezione, del suo gene paralogo SMN2 presenti nel campione. Il numero di copie del gene SMN2 è uno dei fattori che incide sulla variabilità fenotipica delle differenti forme di SMA. La diagnosi prenatale può essere effettuata mediante l'analisi molecolare su amniociti o su villi coriali. Il test molecolare per la SMA mediante analisi MLPA non consente l’identificazione dei portatori o dei malati di SMA nei seguenti casi: 1. mutazioni intrageniche di SMN1: circa il 3,5% dei malati che risultano portatori eterozigoti della delezione di SMN1 hanno piccole delezioni/mutazioni puntiformi, non identificabili con questa metodica, che inattivano la funzionalità del secondo allele (Ogino and Wilson, 2004) 2. duplicazione in Cis di SMN1 (genotipo “2+0”): circa il 2-3% degli individui con 2 copie del gene SMN1 sono in realtà dei portatori eterozigoti della delezione con una probabilità del 50% di trasmetterla alla prole (Ogino and Wilson, 2004) 3. mutazioni de novo: circa il 2% dei pazienti SMA ha una mutazione de novo (Wirth et al, 1997) e, quindi, i genitori risultano avere un dosaggio normale del gene SMN1 4. mosaicismo germinale per la mutazione (Campbell et al, 1998): la presenza di un mosaicismo germinale non permette l’identificazione della condizione di portatore eterozigote nel sangue periferico dei genitori dell’individuo affetto.
Aggiornamento Metodo (2023. AmplideX® SMA Plus Kit in accordo alle indicazioni della casa produttrice). Il kit è basato su amplificazione genica (PCR) ed elettroforesi capillare (CE) su SeqStudio Flex Genetic Analyzers (Thermo Fisher Scientific), per ottenere la quantificazione del numero di copie dell'esone 7 di SMN1. Scopo del test: Il kit AmplideX® SMA Plus è un saggio basato sullamplificazione dellesone 7 dei geni SMN1 e SMN2 insieme ad un controllo endogeno (EC) a partire da DNA genomico purificato: gli ampliconi fluorescenti specifici per SMN1 e SMN2 sono separati tramite elettroforesi capillare e rapportati al controllo endogeno co-amplificato per determinare il numero di copie rispettivo. Il kit permette inoltre di rilevare eventi di conversione genica (SMN1-SMN2 e SMN2-SMN1), la presenza delle varianti c.*3+80T>G e c.*211_*212del di SMN1 associate alla duplicazione genica su singolo cromosoma e della variante c.859G>C di SMN2 correlata ad un fenotipo della malattia meno severo dovuto ad un più efficiente meccanismo di splicing di SMN2. Il numero di copie dellesone 7 di SMN2 e leventuale presenza di geni ibridi (con sequenze appartenenti sia a SMN1 che a SMN2) e delle varianti di SMN1 (c.*3+80T>G and c.*211_*212del) e di SMN2 (c.859G>C) saranno riportati solamente se clinicamente utili a determinare il fenotipo della patologia. Specificità: >99%; Sensibilità: 71-95% (variabile in relazione alletnia) Limiti: Lanalisi è disegnata per eseguire il dosaggio quantitativo dellesone 7 dei geni SMN1 ed SMN2, mentre non è in grado di evidenziare varianti nonsenso, missenso o frameshift della sequenza genica. Circa il 3-8% degli individui portatori sani di Atrofia muscolare spinale presentano due copie del gene SMN1 su un singolo cromosoma e zero copie sullaltro cromosoma; tali soggetti sono definiti portatori silenti o portatori 2+0. Lanalisi quantitativa non è in grado di identificare lo stato di portatore silente. Lo stato di portatore silente può tuttavia essere discriminato dal soggetto con una copia del gene su ciascun cromosoma grazie alla presenza di varianti di singolo nucleotide che si presentano in linkage disequilibrium con la duplicazione genica su singolo cromosoma. La presenza delle varianti di SMN1 c.*3+80 T>G (g.27134 T>G) e c.*211_*212del (g.27706_27707 delAT) suggeriscono la presenza di una duplicazione genica su singolo cromosoma e quindi di un portatore silente. La probabilità di essere un portatore silente varia in base alla presenza/assenza delle suddette varianti in diverse etnie (Luo et al. 2014; Alías et al. 2018). Le due varianti non sono specifiche per il gene SMN1 o il gene SMN2, ma sono rilevate indipendentemente dalla loro presenza su uno o sullaltro gene. I siti di appaiamento dei primers contenuti nel kit AmplideX® SMA Plus non presentano varianti nucleotidiche note con frequenza allelica minore (MAF) superiore allo 0.005, tuttavia la presenza di varianti rare potrebbe alterare il dosaggio quantitativo dei geni SMN1 e SMN2 (Prior et al. 2011).